 Cystinosis is a rare autosomal recessive lysosomal storage disorder caused by mutations in the CTNS gene, which encodes cystinosin — a lysosomal cystine transporter.
Cystinosis is a rare autosomal recessive lysosomal storage disorder caused by mutations in the CTNS gene, which encodes cystinosin — a lysosomal cystine transporter.
Without functional cystinosin, cystine accumulates within lysosomes and forms crystals that damage cells throughout the body.
CTNS encodes, a ubiquitous proton driven lysosomal cystine transporter cystine.
Cystine accumulates within lysosomes in all organs:Impairs lysosomal signaling, defective autophagia, and widespread cellular dysfunction.
There are three clinical forms based on severity and age of onset: Nephropathic (infantile) — the most common and severe form. Symptoms appear in the first year of life.
Intermediate (juvenile) — milder renal involvement, presenting in adolescence.
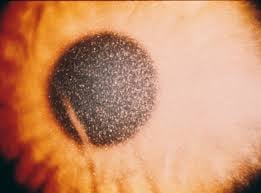
Non-nephropathic (ocular/adult) — limited to photophobia from corneal crystals; no systemic involvement.
Cystine crystals accumulate in virtually every tissue: kidneys, eyes, muscles, liver, thyroid, brain, and more.
The kidney is the earliest and most severely affected organ in cystinosis.
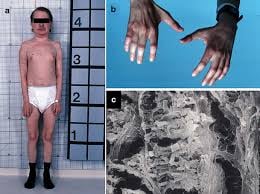
The nephropathic form typically presents with Fanconi syndrome with proximal tubular dysfunction: polyuria, polydipsia, growth failure, electrolyte wasting of phosphate, potassium, sodium, bicarbonate, and rickets.
Without treatment, end-stage renal disease (ESRD) typically occurs by the second decade of life.
Extrarenal complications accumulate over time and include corneal crystals (photophobia, blepharospasm,corneal erosion), hypothyroidism, myopathy that can result in respiratory dysfunction, dysphasia, aspiration pneumonia, diabetes, bone deformities and neurological issues with cognitive decline, and cerebral atrophy.
Non-renal complications of stenosis include photophobia, and corneal erosion, cardiovascular complications, diabetes, hypothyroidism, bone deformities, and fragility, neurologic defects and progressive myopathy that can result in life-threatening respiratory dysfunction, dysphasia, and aspiration pneumonia.
Three allelic forms of Cystinosis exist, with the most severe, and most common is an infantile form.
Renal Fanconi syndrome develops in affected children by 6 to 18 months of age and chronic kidney disease eventually leads to end stage kidney disease.
Intracellular assisting reduction with the cystine depleting agent cysteamine allows assist to exit cells and slow the progression of disease.
To reduce corneal cystine crystal accumulation, cysteamine eye drops are needed every hour during the time the person is awake.
Premature death is inevitable despite these therapies.
Diagnosis Elevated white blood cell (WBC) cystine levels
Slit-lamp examination showing pathognomonic corneal cystine crystals
Genetic testing for CTNS mutations
Treatment Cysteamine (oral) is the cornerstone of systemic treatment.
It depletes lysosomal cystine by forming a mixed disulfide that can exit the lysosome.
It slows progression of kidney disease and prevents/delays extrarenal complications but must be taken lifelong.
Cysteamine eye drops treat corneal crystals specifically.
Supportive care: electrolyte replacement, phosphate supplementation, vitamin D, growth hormone, and management of Fanconi syndrome.
Renal transplantation addresses ESRD but does not prevent extrarenal cystine accumulation, so cysteamine must continue.
Early diagnosis and consistent cysteamine therapy dramatically improve outcomes, delaying ESRD by decades and reducing extrarenal complications.
Neonatal or newborn screening is not yet universal, so diagnosis often comes after symptom onset.
Gene therapy can decrease white cell cyst cystine levels.
Gene therapy with autologous transplantation of CD 34 positive HSPCs transducer with the lentiviral vector containing wild type CTNS complementary DNA.