 Neurofibromatosis type 2 (NF2) is an autosomal dominant genetic disorder characterized by the development of multiple benign tumors in the nervous system, primarily due to mutations in the NF2 gene on chromosome 22q12.
Neurofibromatosis type 2 (NF2) is an autosomal dominant genetic disorder characterized by the development of multiple benign tumors in the nervous system, primarily due to mutations in the NF2 gene on chromosome 22q12.
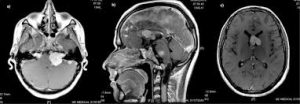
The hallmark of NF2 is the presence of bilateral vestibular schwannomas, which occur in approximately 95% of affected individuals and typically lead to progressive hearing loss, tinnitus, and balance issues.
Neurofibromatosis type II-multiple inherited schwannomas, meningiomas, and ependymomas, is a genetic condition that may be inherited or may arise spontaneously, and causes benign tumors of the brain, spinal cord, and peripheral nerves.
Incidence of the condition is about 1 in 60,000.
NF2 is a life limiting condition.
It is a rare genetic disorder that involves noncancerous tumors of the nerves that transmit balance and sound impulses from the inner ear to the brain.
The types of tumors frequently associated with NF2 include vestibular schwannomas, meningiomas, and ependymomas.
The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII (auditory-vestibular nerve) that transmits sensory information from the inner ear to the brain.
Symptoms depend on the presence, localization and growth of the tumors.
Many people with this condition also experience vision problems.
Neurofibromatosis type II (NF2 or NF II) is caused by mutations of the “Merlin” gene, which seems to influence the form and movement of cells.
The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions.
Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.
NF2 is an inheritable disorder with an autosomal dominant mode of transmission.
There are two forms of the NF2.
The Wishart-phenotype form is characterized by multiple cerebral and spinal lesions in people younger than 20 years and with rapid progression of the tumors.
People with NF2 who develop single central tumours with slow progression after the age of 20 are thought to have the Feiling–Gardner phenotype.
Symptoms can occur at any age, typically in adolescence and early adulthood, and rarely seen in children, and the severity depends on the location of the tumors.
Symptoms include, but are not limited to:
Tinnitus Loss/problems of balance Glaucoma Seizures Hearing loss Vision impairment Numbness or weakness in arms and legs
Hearing loss in those with NF2 almost always occurs after acquisition of verbal language skills, people with NF2 do not always integrate well into Deaf culture and are more likely to resort to auditory assistive technology.
A cochlear implant, can sometimes restore a high level of auditory function even when natural hearing is totally lost.
However, the amount of destruction to the cochlear nerve caused by the typical NF2 schwannoma often precludes the use of such an implant, it an auditory brainstem implant (ABI) can restore some level of hearing, supplemented by lip reading.
NF-2 may be inherited in an autosomal dominant, las well as through random mutation.
It is suspected that one-half of cases are inherited, and one-half are the result of new, de novo mutations.
NF2 is caused by inactivating mutations in the NF2 gene located at 22q12.2 of chromosome 22.
Types of mutations include protein deletions/insertions and nonsense mutations, splice-site mutations, missense mutations, and deletions, in the NH2-terminal domain of merlin proteins have been associated with early tumor onset and poor prognosis in people with NF2
Protein truncating mutations are associated with more severe phenotype.
All people with the condition who have been checked have been found to have some mutation of the same gene on chromosome 22.
The hearing loss caused by NF2 is gradual.
The hearing loss caused by NF2 results from the presences of bilateral cochleovestibular schwannomas (acoustic neuromas), which damage to cochlear nerve causing hearing loss.
Hearing loss may also result from benign tumors that grow on the vestibular and auditory nerves, which lead to the inner ear.
NF2 is caused by a defect in the gene that normally gives rise to Merlin or Schwannomin, located on chromosome 22 band q11-13.1.
Merlin is a structural protein functioning as an actin cytoskeleton regulator.
Merlin regulates multiple proliferative signalling cascades such as receptor tyrosine kinase signalling, p21-activated kinase signalling, Ras signalling, MEK-ERK cascade, MST-YAP cascade.
In a normal cell, the concentrations of active merlin are controlled by processes such as cell adhesion.
Merlin inhibits Rac1 which is crucial for cell motility and tumor invasion.
Merlin deficiency can result in progression through the cell cycle due to the lack of contact-mediated tumor suppression, mainly because of the cell:cell junction disruption, sufficient to result in the tumors characteristic of Neurofibromatosis type II.
Besides its cytoskeletal and cytoplasmic functions Merlin also translocates to the nucleus and suppresses proliferation.
Merlin also plays important role in energy metabolism regulation.
Mutations of NF2 is presumed to result in either a failure to synthesize Merlin or the production of a defective peptide lacking the normal tumor-suppressive effect.
The Schwannomin-peptide consists of 595 amino acids.
Comparison of Schwannomin with other proteins shows similarities to proteins that connect the cytoskeleton to the cell membrane.
Mutations in the Schwannomin-gene alter the movement and shape of affected cells with loss of contact inhibition.
Ependymomas are tumors arising from the ependyma, an epithelium-like tissue of the central nervous system.
Loss of function mutations occurring in chromosome 22q, where Merlin proteins are coded, can promote tumorigenesis, or the creation of new tumorous cells.
Deletions, in the NH2-terminal domain of merlin proteins have been associated with early tumor onset and poor prognosis in affected people.
The so-called acoustic neuroma of NF2 is in fact a schwannoma of the nervus vestibularis, or vestibular schwannoma.
The vestibular schwannomas grow slowly at the inner entrance of the internal auditory meatus, and are derived from the nerve sheaths of the upper part of the nervus vestibularis in the region between the central and peripheral myelin 1 cm from the brainstem.
NF2 is a genetically transmitted condition.
Diagnosis is most common in early adulthood (20–30 years), but can be diagnosed earlier.
NF2 can be diagnosed due to the presence of a bilateral vestibular schwannoma, or an acoustic neuroma, which causes a hearing loss that may begin unilaterally.
If a patient does not meet this criterion of diagnosis, they must have a family history of NF2, and present with a unilateral vestibular schwannoma and other associated tumors (cranial meningioma, cranial nerve schwannoma, spinal meningioma, spinal ependymomas, peripheral nerve tumor, spinal schwannoma, subcutaneous tumor, skin plaque).
More than half of all patients diagnosed with NF2 do not have a family history of the condition.
Peripheral neuropathy, or damage to the peripheral nerves, which can cause causes weakness, numbness and pain in the hands and feet, may also lead to a diagnosis of NF2.
NF2 can present in children with similar symptoms, and causes visual disturbances with cataracts, hamartomas, skin tumors, mononeuropathhy, symptomatic spinal cord tumors, and non-vestibular intracranial tumors.
Diagnostic criteria for NF2:
Bilateral vestibular schwannoma (VS) or family history of NF2 plus Unilateral VS or any two of: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities.
Unilateral VS plus any two of meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
Two or more meningioma plus unilateral VS or any two of glioma, schwannoma and cataract.
Another set of diagnostic criteria:
Detection of bilateral acoustic neuroma by imaging-procedures First degree relative with NF2 and the occurrence of neurofibroma, meningiomas, glioma, or Schwannoma First degree relative with NF2 and the occurrence of juvenile posterior subcapsular cataract.
Treatment
There are several different surgical techniques for the removal of acoustic neuroma, and choice of surgical approach is determined by size of the tumor, hearing capability, and general clinical condition of the person.
Retrosigmoid approach The translabyrinthine approach The middle fossa approach is preferred for small tumors and offers the highest probability of retention of hearing and vestibular function.
Less invasive endoscopic techniques have recovery times are reported to be faster.
Larger tumors can be treated by either the translabyrinthine approach or the retrosigmoid approach.
With large tumors, the chance of hearing preservation is small with any approach.
When hearing is already poor, the translabyrinthine approach may be used.
Auditory canal decompression is another surgical technique that can prolong usable hearing when a vestibular schwannoma has grown too large to remove without damage to the cochlear nerve.
Radiosurgery is a conservative alternative to cranial based surgery.
Radiosurgery can seldom completely destroy a tumor, it can often arrest its growth or reduce its size.
Radiation incurs a higher risk of subsequent malignant change in the irradiated tissues, and this risk is higher in NF2 than in sporadic (non-NF2) lesions.
No medication currently indicated for reduction in tumor burden for NF2 patients.
Bevacizumab has resulted in reduction in tumor growth rates and hearing improvements in some patients.
As hearing loss in individuals with NF2 is generally gradual, eventually profound and sensorineural.
The best options for treatment for hearing loss are cochlear implants and auditory brainstem implants (ABIs), as well as supplementing hearing with lip-reading, cued speech or sign language.
A cochlear implant stimulates the cochlear nerve.
Cochlear implants will work only when the cochleovestibular nerve and the cochlea are still functioning.
Auditory Brainstem Implants are used when the cochlea or any portion of the cochleovestibular nerve are not functioning due to damage to those areas or anatomic abnormalities.
It is an implanted device that sends an electrical signal directly to the cochlear nucleus, allowing sound to bypass the peripheral auditory system and straight into the brain stem.
Prognosis related to early age onset, a higher number of meningiomas and schwannomas and having a decrease in mutation.
Even with an early diagnosis, some patients still die very young.
Meningiomas are tumors that are both intracranial and intraspinal.
Schwannomas are tumors that are often centered on the internal auditory canal.
Patients with NF2 and meningiomas have a higher risk of mortality, and the treatment can be very challenging.
Individuals who develop schwannomas frequently develop hearing loss and deafness and also develop tinnitus after being presented with unilateral hearing loss.
The first symptom that individuals with schwannoma may encounter is dizziness or imbalance.
Missense mutations and large deletions can cause predominantly mild phenotypes.